GenSAS User's Guide
This User's Guide is designed to help you learn how to use GenSAS. The "Using GenSAS" section has very detailed instructions on how to use GenSAS. This section also was written for researchers who have never annotated a genome before and includes some suggestions on best practices. A video tutorial section with brief videos on how to use GenSAS will be coming soon. Please access these sections of the User's Guide by scrolling to the bottom of this page (under the flowcharts), or use the "GenSAS User's Guide" sidebar menu on the right to navigate through the sections of the User's Guide.
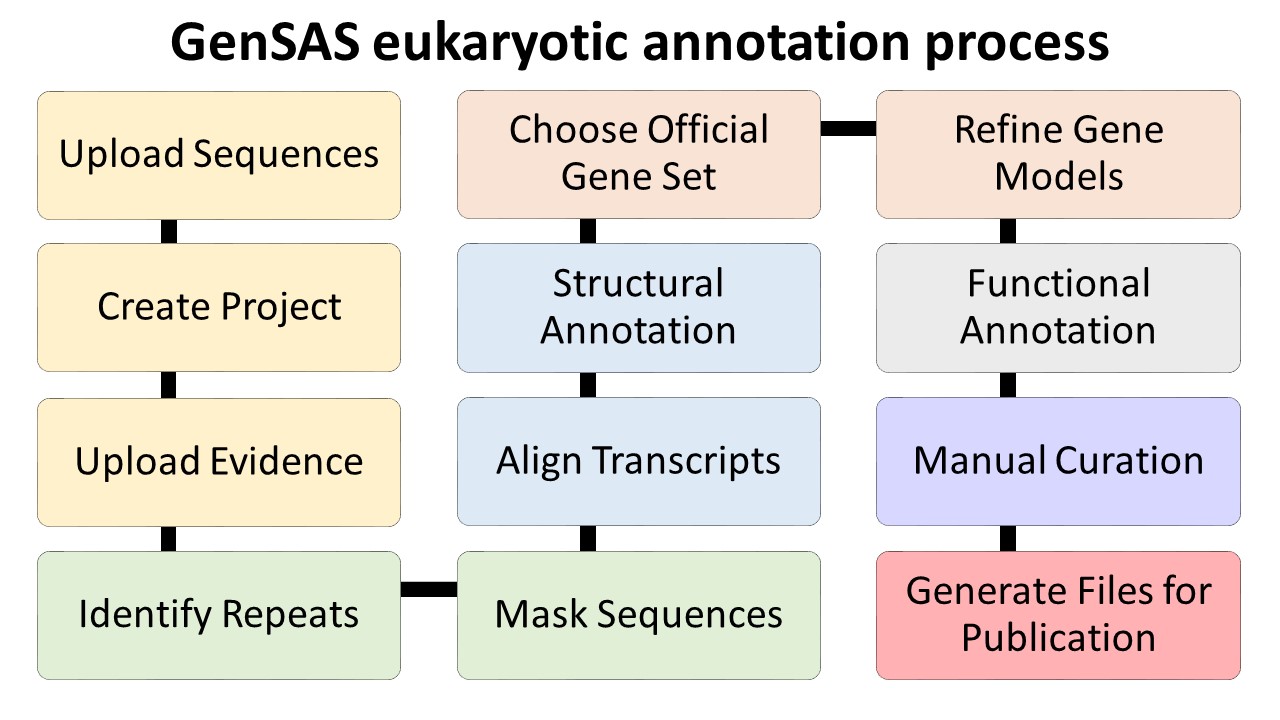
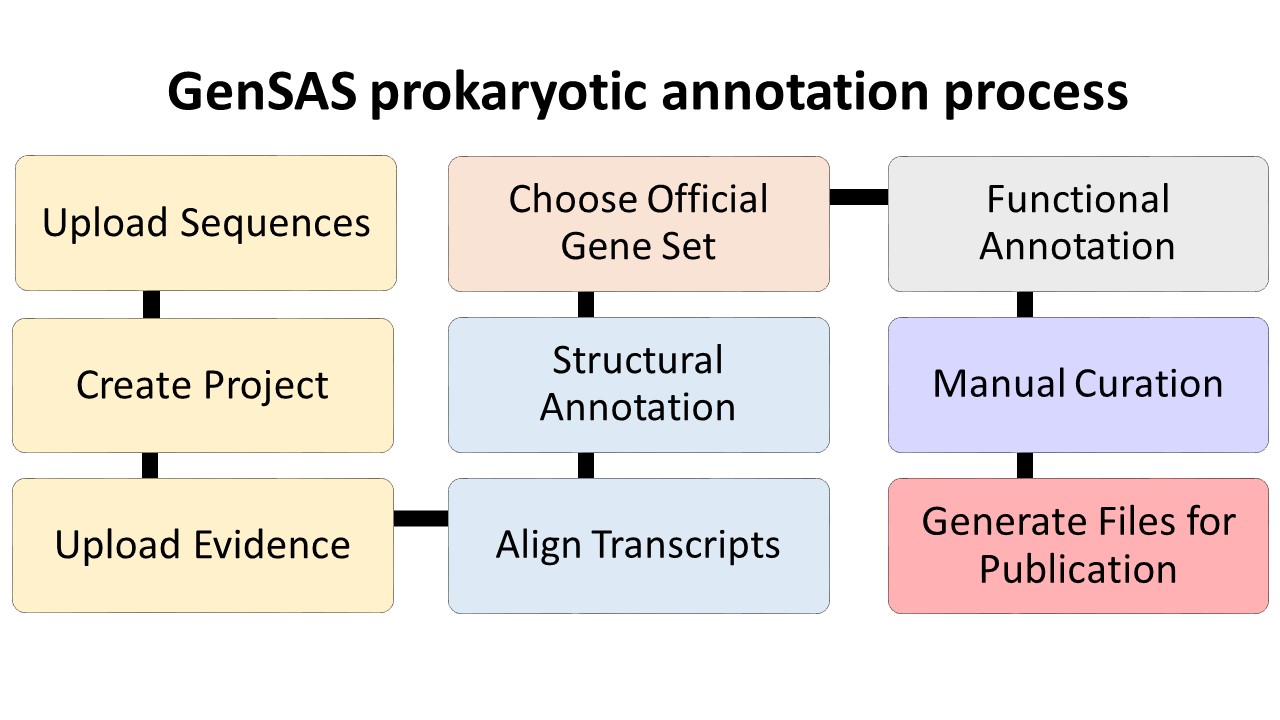
Please make sure that your genome is ready to annotate before loading it into GenSAS. Contigs that are smaller than the average gene size for an organism will not annotate properly with the provided tools. If your genome assembly is predominantly small contigs that are below the average gene size, then your genome probably will not annotate well with GenSAS. If you do not have the metrics of your assembly (i.e. N50, min/max contig size, number of contigs), please consider running the assembly file through a program (such as PRINSEQ) that will provide that data prior to loading it into GenSAS and please note the Project Limits in regards to the number and size of contigs in the assembly. Below are overview diagrams of the annotation process with eukaryotes and prokaryotes.
The steps on the flowcharts with the same color represent the major steps of annotation:
- Project creation/uploading sequences and evidence
- Repeat identification and masking (for eukaryotes)
- Structural annotation
- Selection of official gene set (and refinement for eukaryotes)
- Functional annotation
- Manual curation of the annotation
- Preparing files for publication